Tóm tắt nội dung
Bệnh thalassemia có tỉ lệ gặp phải cao trong các bệnh bẩm sinh. Với những biểu hiện chính là thiếu máu và thừa sắt, cần phải được truyền máu và dùng thuốc thải sắt suốt đời.
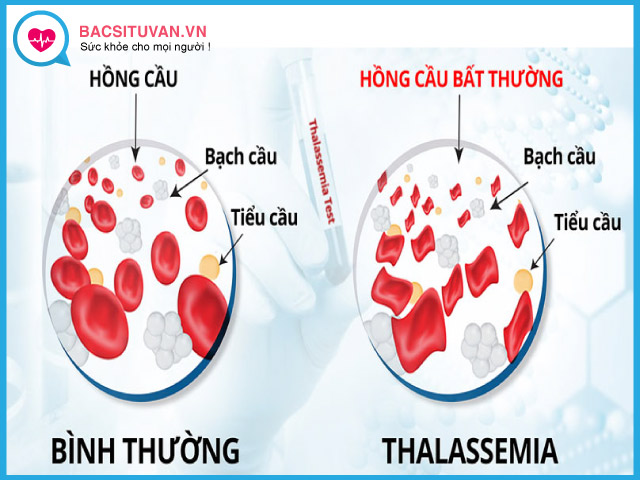
Có hai thể bệnh chính là alpha thalassemia và beta thalassemia, ngoài ra có các thể phối hợp khác như thalassemia và bệnh huyết sắc tố. Mỗi thể bệnh là do bất thường trong tổng hợp một loại chuỗi globin của hồng cầu.
Thalassemia là bệnh di truyền có tỷ lệ cao nhất trên thế giới, theo báo cáo của Liên đoàn Thalassemia Thế giới năm 2012 có khoảng 7% dân số trên thế giới mang gen bệnh huyết sắc tố và thalassemia. Mỗi năm có khoảng 60.000 – 70.000 trẻ em sinh ra bị bệnh thalassemia mức độ nặng. Bệnh tập trung nhiều ở vùng Địa Trung Hải, Trung Đông, Châu Á – Thái Bình Dương trong đó có Việt Nam.
Hiện nay, ở nước ta có trên 12 triệu người mang gen bệnh tan máu bẩm sinh và trên 20.000 người bệnh mức độ nặng cần phải điều trị suốt đời. Mỗi năm có thêm khoảng 8.000 trẻ em sinh ra bị bệnh thalassemia, trong đó có khoảng 2.000 trẻ bị bệnh mức độ nặng và khoảng 800 trẻ không thể ra đời do phù thai.
1. Cấu tạo và vai trò của huyết sắc tố
Thành phần quan trọng nhất của hồng cầu là huyết sắc tố (HST) còn gọi là hemoglobin (Hb) là một protein phức có chứa Fe++, làm nhiệm vụ vận chuyển oxi từ phổi đến tổ chức và vận chuyển CO2 từ tổ chức về phổi, Hb ở trong hồng cầu và chiếm 33% trọng lượng hồng cầu.
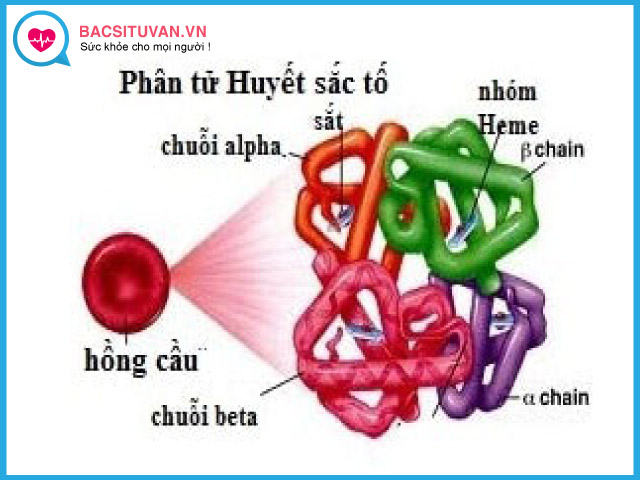
Một phân tử Hb có 4 đơn vị, mỗi đơn vị gồm một chuỗi globin và một nhân hem. Có 5 loại chuỗi globin khác nhau là chuỗi thuộc họ alpha và chuỗi thuộc họ không alpha. Chuỗi thuộc họ không alpha bao gồm chuỗi beta, gamma, delta và epsilon. Các chuỗi kết hợp với nhau theo nguyên tắc giống nhau từng đôi một, trong đó một đôi thuộc họ alpha và một đôi thuộc họ không alpha.
Về cấu trúc không gian thì hai chuỗi giống nhau được xếp đối xứng nhau, 4 chuỗi tạo nên phân tử tựa hình cầu.
Tuỳ theo sự kết hợp các loại chuỗi globin trong phân tử mà có các loại HST hay Hb khác nhau:
- Ở người lớn bình thường chủ yếu là HbA được tạo thành từ 2 chuỗi alpha và 2 chuỗi beta.
- HbA2 gồm 2 chuỗi alpha và 2 chuỗi delta.
- HbF (còn gọi HST bào thai vì chiếm tỷ lệ rất cao ở giai đoạn cuối của thai nhi và trẻ sơ sinh), có cấu tạo gồm 2 chuỗi alpha và 2 chuỗi gamma.
Thành phần HST bình thường theo lứa tuổi:
- Ở trẻ sơ sinh hoặc những thái cuối của thai nhi: HbF chiếm tỷ lệ cao khoảng 60 – 80%, trong khi HbA chỉ chiếm 20 – 40%, HbA2 chiếm tỷ lệ rất thấp chỉ 0,03 – 0,6%.
- Ở trẻ trên 5 tuổi và người trưởng thành: chủ yếu là HbA chiếm đến 96 – 98%, HbA2 chiếm khoảng 2 – 3 %, trong HbF chỉ chiếm 0,4 – 1%.
2. Nguyên nhân gây bệnh thalassemia

Thalassemia là bệnh di truyền gen lặn trên nhiễm sắc thể thường, do vậy tỷ lệ nam và nữ bị bệnh như nhau.
Nếu cả bố và mẹ mang gen bệnh (nhưng không bị bệnh) thì mỗi lần sinh có 25% nguy cơ con bị bệnh, 50% khả năng con mang một gen bệnh và 25% khả năng con bình thường.
Nếu chỉ có bố hoặc mẹ mang gen bệnh, thì trong mỗi lần sinh con thì có 50% nguy cơ con có gen bệnh (nhưng không bị bệnh).
Khi gen globin bị tổn thương, chuỗi globin đó không được tổng hợp hay tổng hợp bị giảm nên thiếu huyết sắc tố gây thiếu máu.
Căn nguyên gây bệnh chính là thừa tương đối chuỗi globin tương ứng. Các chuỗi globin thừa này sẽ trùng hợp tạo nên các thể vùi huyết sắc tố. Những thể vùi này không có tác dụng vận chuyển oxi và gắn lên màng hồng cầu làm thay đổi tính thấm, tính mềm dẻo của màng hồng cầu làm hồng cầu dễ vỡ. Hồng cầu vỡ gây thiếu máu, giải phóng sắt gây thừa sắt và các biểu hiện lâm sàng.
3. Phân loại mức độ bệnh thalassemia
Bệnh tan máu bẩm sinh có nhiều mức độ lâm sàng khác nhau:
- Mức độ rất nặng: thai nhi phù, thường tử vong trước hoặc ngay khi sinh
- Mức độ nặng và trung bình: có các biểu hiện lâm sàng chung như thiếu máu (trung bình đến nặng), vàng da, gan lách to, chậm phát triển thể chất, biến dạng xương (xương sọ, xương mặt, …), rối loạn nội tiết như đái đường, suy giảm chức năng sinh dục…
- Mức độ nhẹ: chỉ bị thiếu máu nhẹ, dễ bị chẩn đoán nhầm với các bệnh lý khác như thiếu máu thiếu sắt.
Người mang gen bệnh tan máu bẩm sinh không có biểu hiện lâm sàng, có thể không thiếu máu.
4. Triệu chứng bệnh thalassemia
Do mất cân bằng trong quá trình tổng hợp chuỗi globin gây ra các hậu quả:
Thiếu máu
Là tình trạng thiếu máu mạn tính trong suốt cuộc đời người bệnh. Nguyên nhân do các chuỗi globin thừa lắng đọng trong các tế bào đầu dòng hồng làm quá trình sinh hồng cầu không hiệu lực từ trong tủy xương, hồng cầu trưởng thành dễ vỡ, bị tiêu hủy sớm hơn ở lách và lượng huyết sắc tố trong mỗi hồng cầu thấp. Tất cả các nguyên nhân này dẫn đến lượng huyết sắc tố của bệnh thalassemia thấp hơn bình thường.
Thay đổi cấu trúc xương
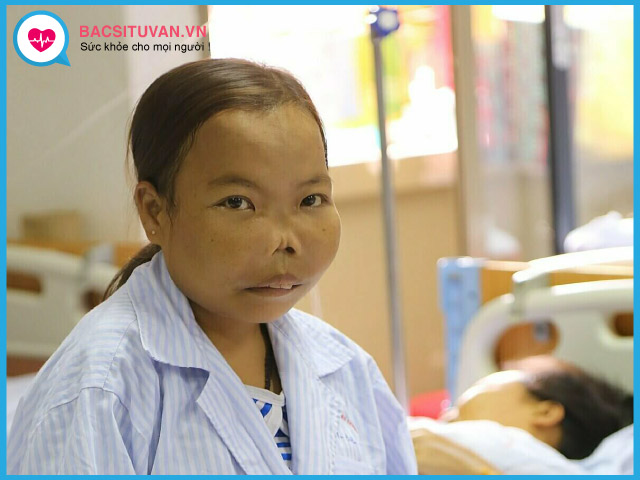
Do thiếu máu, cơ thể phản ứng bằng tăng sinh hồng cầu, mở rộng diện tích sinh hồng cầu trong tuỷ xương dẫn đến thay đổi cấu trúc xương sọ, mặt và đầu xốp các xương dài. Một số trường hợp có các u sinh máu trong ống tuỷ, phổi... Điều này làm gương mặt người bệnh thalassemia bị biến dạng: trán dô, mũi tẹt, gò má cao, răng vẩu, xương dễ gãy, giảm mật độ xương, loãng xương.
Lách to
Chuỗi globin thừa tạo thành thể vùi trong hồng cầu làm màng hồng cầu mất độ mềm mại, dễ bị bắt giữ tại lách, làm lách phì đại, với một số lượng lớn hồng cầu được giữ trong lách làm giảm lượng hồng cầu trong máu do đó càng làm máu bị loãng hơn. Nếu đã cắt lách thì hiện tượng này sẽ xảy ra đối với gan.
Rối loạn chuyển hóa sắt
Tuỷ xương tăng sinh hồng cầu kích thích cơ thể tăng hấp thu sắt từ đường tiêu hóa, bên cạnh đó do thường xuyên phải truyền khối hồng cầu nên tổng lượng sắt của cơ thể tăng cao nhanh chóng. Khi sắt huyết thanh tăng 10 – 15 lần, các vị trí gắn sắt của transferrin đã bão hoà hết, sắt sẽ gắn không đặc hiệu với các chất khác như albumin, citrate, aminoacid và được lắng đọng tại các tổ chức như gan, tim, tuyến nội tiết làm tổn thương các cơ quan này. Điều này dẫn đến tình trạng xơ gan, suy gan, suy tim, suy các tuyến yên tuyến sinh dục, tiểu đường, suy giáp, suy cận giáp…
Rối loạn đông cầm máu
Người bệnh tan máu bẩm sinh có những biến đổi về đông cầm máu, nhìn chung có xu hướng tăng đông máu.
5. Điều trị bệnh thalassemia
Hai biện pháp chính điều trị bệnh tan máu bẩm sinh hiện nay là truyền máu và thải sắt. Bên cạnh đó, một số biện pháp phổ biến khác cũng được sử dụng cho điều trị.
Truyền máu
Do bị thiếu máu mạn tính, cần phải truyền máu định kỳ, suốt cả cuộc đời. Khoảng cách giữa các lần truyền máu là 2 – 5 tuần. Chế phẩm sử dụng là khối hồng cầu.
Thải sắt
Mục đích để chống thừa sắt, nhằm đưa nồng độ sắt trong cơ thể về giới hạn bình thường. Thường phải duy trì dùng thuốc thải sắt trong suốt cuộc đời.
Cắt lách
Được chỉ định khi có tăng nhu cầu truyền máu hơn 50% so với ban đầu trong 6 tháng, lách quá to gây đau, giảm bạch cầu hoặc giảm tiểu cầu nặng (do cường lách).
Ghép tế bào gốc
Là phương pháp điều trị hiện đại, có thể chữa khỏi bệnh thalassemia, tuy nhiên chi phí điều trị khá tốn kém. Hơn nữa nếu đã bị nhiễm sắt nặng tại gan thì tỷ lệ thành công thấp.
- Chăm sóc toàn diện: Để phòng ngừa và hạn chế các biến chứng, nâng cao chất lượng cuộc sống.
- Điều trị biến chứng: Tùy theo biểu hiện, điều trị biến chứng như suy tuyến nội tiết, tiểu đường, suy tim, xơ gan, loãng xương, rối loạn đông máu…
6. Một số điểm lưu ý chăm sóc người bệnh thalassemia
Người bệnh tan máu bẩm sinh thường bị quá tải sắt, sắt dư thừa tích tụ trong gan, tim, tinh hoàn/ buồng trứng, tuyến yên. Từ đó làm tổn thương, suy giảm chức năng của các cơ quan này.

Người bệnh tan máu bẩm sinh nên lựa chọn các loại thực phẩm cho năng lượng cao mà chứa hàm lượng sắt thấp:
- Hạn chế các thức ăn có chứa hàm lượng sắt cao như: thịt bò, thịt trâu, thịt gà chọi, thịt chó, tim, gan… Các loại rau có màu xanh đậm như: rau bina, cải xoong, rau ngót, rau muống, rau dền …Các loại nấm.
- Nên uống nước chè tươi hàng ngày và ngay sau bữa ăn để làm giảm hấp thu sắt từ thực phẩm.
- Hạn chế các chất kích thích và nước ngọt có ga: rượu, bia, café, coca…
- Nên bổ sung các loại vitamin, khoáng chất cần thiết cho sự phát triển của xương nhằm duy trì thể trạng và không làm trầm trọng thêm vấn đề quá tải sắt.
- Bổ sung các thực phẩm chứa nhiều canxi, kẽm và vitamin D để cho xương vững chắc như tôm, cua, cá…
- Hạn chế muối trong khẩu phần ăn hằng ngày, lượng muối từ 4 – 6g/ ngày.
Chế độ sinh hoạt đối với người mắc bệnh thalassemia:
Người bệnh tan máu bẩm sinh có thể sinh hoạt bình thường, tuy nhiên hạn chế lao động nặng hoặc các hoạt động gắng sức.
Tránh nhiễm trùng:
- Giữ gìn vệ sinh sạch sẽ, rửa tay thường xuyên, tiêm vắc xin phòng bệnh đầy đủ
- Giữ ấm cơ thể khi trời lạnh, đảm bảo vệ sinh an toàn thực phẩm.
Tập thể dục thường xuyên, chọn các bài tập nhẹ nhàng, phù hợp với lứa tuổi và tình trạng bệnh.
7. Dự phòng bệnh thalassemia

Hai người khỏe mạnh có thể cùng mang gen bệnh và khi kết hôn có nguy cơ sinh ra con bị bệnh thể nặng. Vì thế các biện pháp phòng tránh bệnh tan máu bẩm sinh:
- Các bạn trẻ và những người trong độ tuổi sinh đẻ nên chủ động xét nghiệm, tầm soát gen bệnh càng sớm càng tốt.
- Người mang gen bệnh cần được tư vấn và quản lý nguồn gen để tránh sinh ra con bị bệnh thể nặng.
- Các cặp vợ chồng cùng mang gen bệnh đã kết hôn cần được tư vấn trước khi mang thai và thực hiện các biện pháp chẩn đoán sàng lọc phù hợp.
- Nếu người vợ đã mang thai cần sàng lọc trước sinh trong những tháng đầu nhằm phát hiện gen bệnh có thể có ở thai nhi và tư vấn, đình chỉ nếu phát hiện thai nhi bị bệnh thalassemia mức độ nặng.
Bệnh Thalassemia


