Tóm tắt nội dung
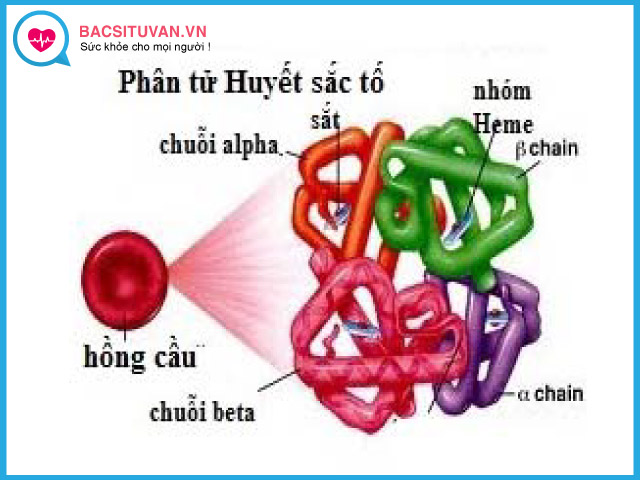
Hemoglobin được tạo từ phần nhân hem (chứa sắt) và phần globin. Globin bao gồm các chuỗi acid amin, các chuỗi globin khác nhau được đặt tên là alpha, beta, delta và gamma.
Ở người trưởng thành, chủ yếu là hemoglobin A (HbA) chiếm đến 97%, trong đó phần globin gồm 2 chuỗi alpha và 2 chuỗi beta. Ở thời kì bào thai và sơ sinh thì chủ yếu là hemoglobin F (HbF) globin gồm 2 chuổi alpha và 2 chuỗi gamma.
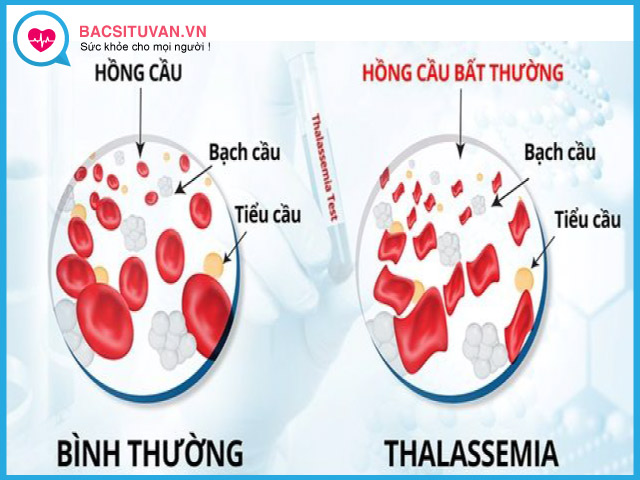
Bệnh thalassemia hay còn gọi là bệnh tan máu bẩm sinh, bệnh thiếu máu do tan máu bẩm sinh… đây là bệnh di truyền gen lặn trên nhiễm sắc thể thường. Bệnh xảy ra do khiếm khuyết tổng hợp 1 hay nhiều chuỗi globin. Rối loạn này dẫn đến một số lượng lớn các tế bào hồng cầu bị phá hủy gây thiếu máu.
1. Nguyên nhân gây bệnh beta thalassemia
Bệnh thalassemia được gọi tên theo chuỗi globin bị khiếm khuyết, gồm hai loại chính là alpha thalassemia và beta thalassemia.
-
Alpha thalassemia gồm 4 gen nằm trên nhiễm sắc thể 16, đột biến alpha thalassemia được gây ra bởi việc mất hoặc đột biến ở một hoặc nhiều trong bốn gen alpha globin. Đột biến gây ra sự giảm sản xuất alpha globin. Càng nhiều gen bị ảnh hưởng, cơ thể càng ít tạo ra alpha globin, bệnh càng nặng.
-
Beta thalassemia gồm 2 gen nằm trên nhiễm sắc thể 11, đột biến beta thalassemia được gây ra bởi đột biến ở một hoặc cả hai gen beta globin. Mức độ nghiêm trọng của bệnh thiếu máu do beta thalassemia phụ thuộc vào đột biến xuất hiện và sự giảm sản xuất beta globin.
Có nhiều yếu tố làm tăng nguy cơ mắc bệnh thiếu máu do beta thalassemiachẳng hạn như:
- Có người thân sinh sống trong khu vực có các đột biến cụ thể của gen locus Beta thalassemia, đặc biệt là Đông Nam Á
- Bố, mẹ bị bệnh hoặc có anh chị ruột đã được phát hiện là bị bệnh
- Được chẩn đoán bệnh beta thalassemia trong thai kỳ trước đó….
2. Chẩn đoán bệnh thiếu máu do beta thalassemia
Chẩn đoán bệnh thiếu máu do thalassemia dựa vào các triệu chứng như:
Yếu tố gia đình: ông bà (nội, ngoại), bố mẹ, anh chị ruột… đã được xác định là bị bệnh hoặc phát hiện mang gen bệnh
Biểu hiện lâm sàng
Do mất cân bằng trong quá trình tổng hợp chuỗi globin nên dẫn đến việc sinh hồng cầu không đáp ứng được nhu cầu của cơ thể và gây ra các hậu quả:
Thiếu máu
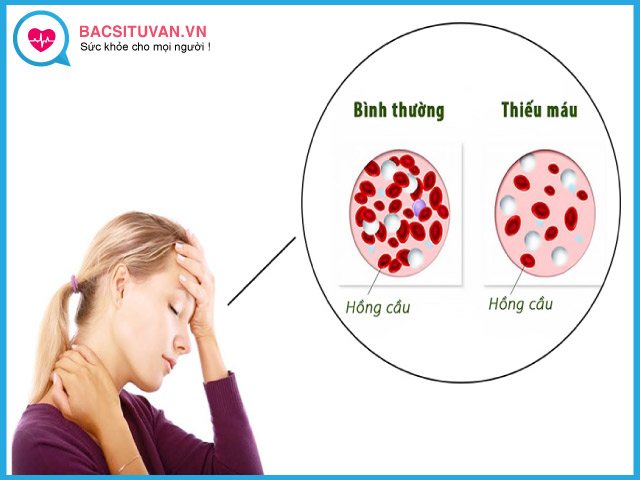
Thiếu máu kéo dài suốt đời do các chuỗi globin thừa lắng đọng trong các tế bào đầu dòng hồng làm quá trình sinh hồng cầu không hiệu lực từ trong tủy xương. Hồng cầu trưởng thành bị tiêu hủy sớm hơn ở lách và lượng huyết sắc tố trong mỗi hồng cầu thấp. Tất cả các nguyên nhân này dẫn đến lượng huyết sắc tố thấp hơn bình thường.
Thay đổi cấu trúc xương
Do thiếu máu, cơ thể phản ứng bằng tăng sinh hồng cầu, mở rộng diện tích sinh hồng cầu trong tuỷ xương dẫn đến thay đổi cấu trúc xương sọ, mặt và đầu xốp các xương dài, một số trường hợp có các u sinh máu. Điều này làm gương mặt bị biến dạng như trán dô, mũi tẹt, gò má cao, răng vẩu, xương dễ gãy, giảm mật độ xương, loãng xương.
Lách to
Chuỗi globin thừa tạo thành thể vùi trong hồng cầu làm hồng cầu mất độ mềm mại, dễ bị bắt giữ tại lách, làm lách phì đại, với một số lượng lớn hồng cầu được giữ trong lách làm giảm lượng hồng cầu trong máu do đó càng làm máu bị loãng hơn. Nếu lách bị cắt thì hiện tượng này sẽ xảy ra đối với gan.
Rối loạn chuyển hóa sắt

Tuỷ xương tăng sinh hồng cầu kích thích cơ thể tăng hấp thu sắt từ đường tiêu hóa, bên cạnh đó do thường xuyên phải truyền khối hồng cầu để điều trị thiếu máu nên tổng lượng sắt của cơ thể tăng cao nhanh chóng.
Khi sắt huyết thanh tăng 10 – 15 lần, các vị trí gắn sắt của transferrin đã bão hoà hết, sắt sẽ gắn không đặc hiệu với các chất khác như albumin, citrate, acid amin và được lắng đọng tại các tổ chức như gan tim, tuyến nội tiết làm tổn thương các cơ quan này. Điều này dẫn đến bệnh nhân bị xơ gan, suy gan, suy tim, suy các tuyến yên tuyến sinh dục, tiểu đường đường, suy giáp, suy cận giáp…
Rối loạn đông cầm máu
Người bệnh tan máu bẩm sinh có những biến đổi về đông cầm máu, nhìn chung có xu hướng tăng đông.
Xét nghiệm huyết đồ
Đây là xét nghiệm đầu tiên được các bác sĩ chỉ định để chẩn đoán tình trạng thiếu máu. Xét nghiệm này rất quan trọng đặc biệt đối với các tuyến y tế cơ sở trong bước đầu chẩn đoán bệnh.
- Số lượng hồng cầu giảm, số lượng huyết sắc tố giảm: tiêu chuẩn để chẩn đoán thiếu máu.
- Các chỉ số khác của hồng cầu: MCV (thể tích khối hồng cầu), MCH (lượng huyết sắc tố trung bình hồng cầu) và MCHC (nồng độ huyết sắc tố trung bình hồng cầu) đều thấp (thể hiện một thiếu máu nhược sắc thể tích hồng cầu nhỏ).
- Hồng cầu lưới máu ngoại vi tăng: khả năng đáp ứng của tủy xương khi có tình trạng thiếu máu.
- Tăng hồng cầu nhiễm sắt.
- Sức bền hồng cầu thường tăng.
- Xuất hiện nhiều hình dạng hồng cầu bất thường: hồng cầu hình giọt nước, hồng cầu hình bia bắn, hồng cầu hình cầu, các mảnh vỡ hồng cầu, hồng cầu hình răng cưa, hồng cầu có chấm ưa base,… và có thể có xuất hiện hồng cầu non trong máu ngoại vi.
Xét nghiệm tủy đồ
Tế bào tủy tăng sinh mạnh dòng hồng cầu, xuất hiện nhiều giai đoạn của tế bào hồng cầu.
Điện di huyết sắc tố
Xét nghiệm này giúp xác định thành phần các loại hemoglobin (tiểu đơn vị vận chuyển oxy) có trong máu.
- Đây là xét nghiệm quan trọng giúp định hướng chẩn đoán bệnh thiếu máu tan máu bẩm sinh ở những trường hợp bác sĩ nghi ngờ.
- Xét nghiệm giúp theo dõi hiệu quả điều trị ở những người mắc bệnh bất thường huyết sắc tố nói chung.
- Xét nghiệm hỗ trợ xác định khả năng di truyền cho thế hệ sau ở những cặp vợ chồng mang gen bệnh mà có mong muốn sinh con.
Ở người trưởng thành khỏe mạnh, giá trị bình thường của kết quả điện di là:
- HbA: 95% - 98%.
- HbA2: 2% - 3%.
- HbF: 0.8% - 2%.
- HbS: 0%.
- HbC: 0%.
Trẻ sơ sinh:
- HbA: 20 - 40%.
- HbA2: 0.03 - 0.6%.
- HbF: 60 - 80%.
Bệnh Thalassemia xuất hiện huyết sắc tố bất thường:
- HbF tăng: β - thalassemia tăng HbF.
- HbA2 tăng: β - thalassemia tăng HbA2.
- HbE tăng: bệnh huyết sắc tố E.
- HbH tăng: α - thalassemia tăng HbH.
Xác định ADN
Đây là xét nghiệm giúp chẩn đoán xác định bệnh. Xét nghiệm phát hiện bất thường vật chất di truyền ở những người mang gen bệnh thalassemia.
Các xét nghiệm sinh hóa kết hợp
- Bilirubin toàn phần tăng, chủ yếu tăng gián tiếp.
- Sắt huyết thanh tăng, ferritin tăng.
3. Điều trị thiếu máu do beta thalassemia
Biện pháp điều trị chủ yếu là truyền máu để chống thiếu máu và tăng thải sắt. Bên cạnh đó, một số biện pháp phổ biến khác cũng được sử dụng cho điều trị bệnh.
Truyền máu
Do bị thiếu máu mạn tính, cần phải truyền máu định kỳ, suốt cả cuộc đời. Khoảng cách giữa các lần truyền máu là 2 – 5 tuần. Chế phẩm sử dụng là khối hồng cầu.
Thải sắt
Mục đích để chống quá tải sắt ở người bệnh, nhằm đưa nồng độ sắt trong cơ thể về giới hạn bình thường. Thường phải duy trì dùng thuốc thải sắt trong suốt cuộc đời.
Cắt lách
Được chỉ định khi có tăng nhu cầu truyền máu hơn 50% so với ban đầu trong 6 tháng, lách quá to gây đau hoặc giảm bạch cầu, giảm tiểu cầu nặng (do cường lách).
Ghép tế bào gốc
Là phương pháp điều trị hiện đại, có thể chữa khỏi bệnh tan máu bẩm sinh. Tuy nhiên, chi phí điều trị khá tốn kém. Ghép tế bào gốc được chỉ định đối với trường hợp thalassemia mức độ nặng, dưới 16 tuổi, chưa có quá tải sắt mức độ nặng và có người cho tế bào gốc phù hợp HLA.
- Chăm sóc toàn diện: để phòng ngừa và hạn chế các biến chứng, nâng cao chất lượng cuộc sống cho người bệnh.
- Điều trị biến chứng: tùy theo biểu hiện, điều trị biến chứng như suy tuyến nội tiết, tiểu đường, suy tim, xơ gan, loãng xương, rối loạn đông máu…
4. Dự phòng bệnh thiếu máu do beta thalassemia

Bệnh thalassemia là bệnh di truyền gen lặn trên NST thường, nên hai người khỏe mạnh có thể cùng mang gen bệnh (gen lặn). Nếu hai người mang gen bệnh mà kết hôn có nguy cơ sinh ra con bị bệnh thể.
Vì thế, các biện pháp phòng bệnh thalassemia tốt nhất là sàng lọc trước hôn nhân để phòng bệnh ở thế hệ sau.
Các biện pháp phòng tránh bệnh tập trung vào:
- Những người ở độ tuổi sinh đẻ, nên chủ động nên chủ động xét nghiệm tầm soát gen bệnh càng sớm càng tốt.
- Người mang gen bệnh cần được tư vấn và quản lý nguồn gen để tránh sinh ra con bị bệnh, đặc biệt là tránh sinh ra con bị bệnh nặng.
- Nếu các cặp vợ chồng cùng mang gen bệnh thì cần được tư vấn trước khi mang thai và thực hiện các biện pháp chẩn đoán sàng lọc trước sinh phù hợp.
- Nếu người vợ của cặp vợ chồng đều mang gen bệnh mà đã mang thai cần sàng lọc trước sinh trong những tháng đầu nhằm phát hiện gen bệnh có thể có ở thai nhi và tư vấn, đình chỉ nếu phát hiện thai nhi bị bệnh tan máu bẩm sinh mức độ nặng.
Thiếu máu


